

After I took Medical Device Software Process Blackbelt course in Udemy, I decided to write this article.
There are two types of medical devices software; SiMD and SaMD. SiMD stands for Software in a medical device and SaMD stands for Software as a medical device. SiMD can be found in implantable medical devices such as pacemaker and cardiovascular electronic devices. SiMD is the firmware in general. SaMD is without being a part of hardware. It can be found in mobile application which retrieves data from implantable medical devices or software which keep track of patient's health chart.
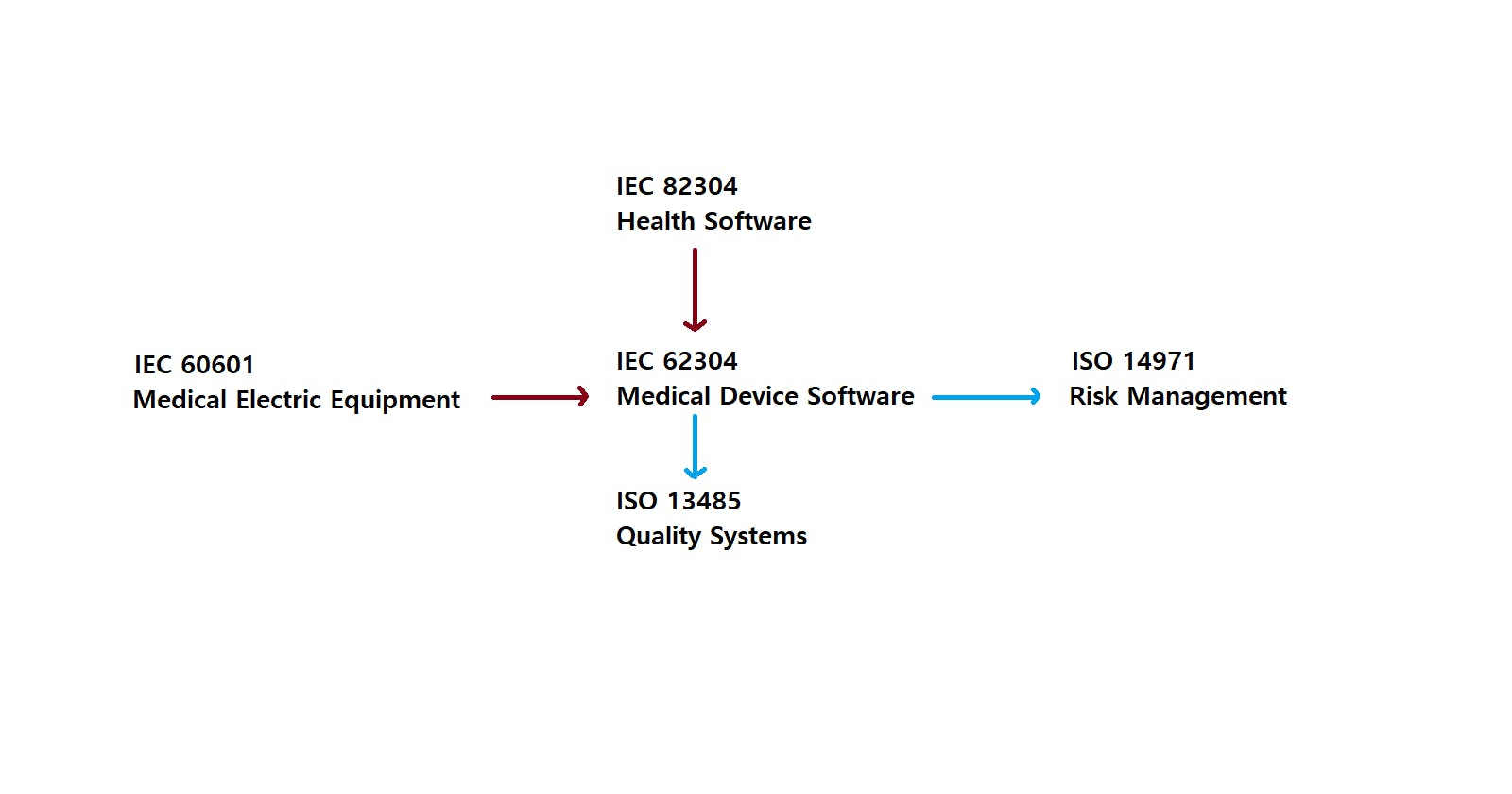
Each medical devices software uses the different international standards to comply with. IEC 82304 is for SaMD and IEC 62304 is for SiMD. However, when you develop the medical devices which include software, there are more standards to consider such as ISO 14971 for risk management and ISO 13485 for quality management.
However, not all of medical devices require all the same amount of documentations. How to classify medical devices? Based on where you live and where that device will be used, classification could be slightly varied. FDA defines three classes; Class I, Class II and Class III. Class I: A medical device with low to moderate risk that requires general controls. Class II: A medical device with a moderate to high risk that requires special controls. Class III: A medical device with high risk that requires premarket approval. Similarly, in IEC 62304, there are Class A, Class B and Class C where it is divided up by the risk level. The number of required amount of documentation will be Class I < Class II < Class III and Class A < Class B < Class C.
Software development lifecycle workflow is defined in IEC 62304 below:
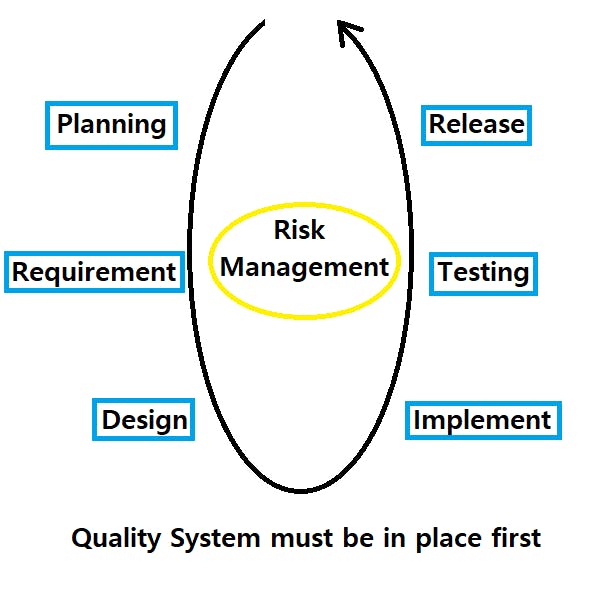
Software Development Lifecycle Summary:
Sub-clause 5.1.1 section c: "The [Software development] plan shall address the following ... traceability between system requirements, software requirements, software system test and risk control measures implemented in software"
Sub-clause 5.2.2 says software requirements should include "...functional and capability, system inputs and outputs, interfaces, alarms, warnings, and operator messages, security, usability, data definition and database, installation and acceptance, methods of operation and maintenance, user documentation, maintenance, and regulatory..."
Sub-clause 5.3.1: "The manufacturer shall transform the requirements for the medical device software into a documented architecture"
Sub-clause 5.4.2: "The manufacturer shall document a design with enough detail to allow correct implementation of each software unit.
Sub-clause 5.4.3 asks: "Does the software code implement requirements including risk control measures?"
Sub-clause 5.6.4 says: "The manufacturer shall address whether the integrated software item performs as intended"
Sub-clauses 5.7.1 says: "The manufacturer shall perform a test of tests ... such that all software requirements are covered"
Sub-clauses 5.8.1 says "The manufacturer shall ensure the software verification has been completed ... before the software is released"\